Amid the skyrocketing cost of clinical trials, the increase in collection and use of digital data, and the FDA’s commitment to considering real-world data (RWD) in its regulatory process, a growing number of life-sciences companies are looking at and employing external or synthetic control arms.
External or synthetic control arms can eliminate the need for placebo controls and incorporate cutting-edge technologies such as AI to unlock the full potential of RWD to evaluate the comparative effectiveness of a therapy.
Arnaub Chatterjee, senior VP of product, Medidata Acorn AI, says external control arms yield several benefits throughout the trial process, especially in patient recruitment and improving trial efficiency.
 “Clinical trials often present an opportunity for patients to receive certain treatments they would not have access to otherwise," Mr. Chatterjee says. “However, patients are often reluctant to engage in clinical trials out of fear of being placed in a control group and not getting the experimental therapy, given that they need lifesaving treatments now. In fact, 9 out of 10 clinical trials worldwide can’t recruit enough people within their target time frames, and some fail altogether due to lack of participants. External control arms solve this issue by ensuring that more patients who choose to participate in the trial can receive the experimental therapy."
“Clinical trials often present an opportunity for patients to receive certain treatments they would not have access to otherwise," Mr. Chatterjee says. “However, patients are often reluctant to engage in clinical trials out of fear of being placed in a control group and not getting the experimental therapy, given that they need lifesaving treatments now. In fact, 9 out of 10 clinical trials worldwide can’t recruit enough people within their target time frames, and some fail altogether due to lack of participants. External control arms solve this issue by ensuring that more patients who choose to participate in the trial can receive the experimental therapy."
Terri Madison, Ph.D., senior VP and general manager of Certara’s evidence and access team, says the obvious advantage of an external control arm is to provide a comparator for a single-arm trial where there is otherwise no ethical choice. This includes circumstances in which there is no approved product that can be used as a comparator product, or where the trial’s endpoint is survival so it may be unethical to give participants a placebo. “For example, our model-based meta-analysis work supported the accelerated approval of blinatumomab in adult patients with relapsed/refractory acute lymphoblastic leukemia based on a single-arm trial with 185 patients, where we evaluated the effect of blinatumomab relative to existing salvage therapies for complete remission, duration of complete remission, and overall survival using meta-analysis models and clinical trial simulations," she says.
External control arms can also lower trial costs by reducing the number of patients needing active management throughout the trial and expediting access to experimental treatment for patients. “These benefits are especially apparent in trials for rare diseases, where the potential patient population is incredibly small and assembling a control group is very difficult," Mr. Chatterjee says.
“Perhaps a less obvious advantage is the ability to provide timely and cost-effective evidence to streamline the evidence needed to determine the safety and efficacy of a product aimed to treat a rare disease with high unmet need," Dr. Madison says. “There are well-published examples of this, such as avelumab for Merkel cell carcinoma, but don’t forget there are unpublished examples where the external control demonstrated the product was not better than current or historical standard of care, an equally important finding.
“Also less publicized, but well-established, is the use of external controls to evaluate real-world product safety," she continues. “For years, regulators have required post-authorization non-interventional studies to assess real-world safety through the conduct of exposure-based registries. Well-matched external controls are essential to contextualize results observed from these exposure-based registries."
Cautious Optimism
 The concept of an external control arm is not new. The National Center for Biotechnology Information of the U.S. National Library of Medicine identified 22 submissions made to the FDA as of May 2020 that used external data and synthetic control methods to establish clinical efficacy, with more than half occurring in the previous two years alone. Thirteen of these submissions used published randomized control trial data for their external controls, and six used observational data.
The concept of an external control arm is not new. The National Center for Biotechnology Information of the U.S. National Library of Medicine identified 22 submissions made to the FDA as of May 2020 that used external data and synthetic control methods to establish clinical efficacy, with more than half occurring in the previous two years alone. Thirteen of these submissions used published randomized control trial data for their external controls, and six used observational data.
According to Dr. Madison, ICH-E10, which covers choice of control group in European clinical trials, and the corresponding FDA guidance have addressed use of external controls since 2000. And there are several examples where FDA has approved products based on single-arm trials, supplemented with external comparator information to contextualize the single-arm trial results.
“The principles outlined in the FDA guidance from May 2001 have not really changed," she notes. “FDA is accepting of ECA evidence for serious diseases with high unmet need where the outcomes are important, objective, and predictable, for example survival, if no internal comparator arm is possible or feasible."
What is new is the term “synthetic control." Although some say it is interchangeable with “external control," Dr. Madison cautions it may not be interchangeable in the FDA’s thinking.
“Synthetic can imply ‘artificially generated data,’ even if modeled or based on real data," she says. “To date, the most common applications accepted by the FDA that had an ECA have used natural history information obtained from medical records. Based on current precedent and guidance documents, it seems that as the model complexity in generating a synthetic control arm increases, the less likely it is that FDA would accept such data as a control arm to a pivotal study when making authorization decisions."
Dr. Madison says Certara’s Clinical Outcomes Database Explorer (CODEx) interface can be used to access its clinical trial databases, currently available in more than 50 indications. “Using our clients’ proprietary patient-level data, plus CODEx summary-level trial data, we can create synthetic control cohorts that are comparable to their single-arm trial population using model-based meta-analysis techniques," she says. “Certara is also developing external control arms using patient-level RWD for supporting regulatory approvals as well as securing market access."
Medidata’s Synthetic Control Arm (SCA) offering pulls data from Medidata’s pool of 7 million anonymized patient records across 25,000 trials. According to Mr. Chatterjee, it is the only synthetic control group created with cross-industry historical trial data.
Medidata first used its SCA in 2019 when collaborating with Friends of Cancer Research to study whether historical clinical trial data in its SCA could replicate the same results as a standard arm in non-small cell lung cancer (NSCLC) trials.
“For this case study, we pulled historical clinical trial data from Stage III/IV NSCLC patients who received platinum-based chemotherapy treatment to develop a synthetic control group, which yielded similar conclusions to those of traditional control methods and showed synthetic control models are viable options for upholding rigorous drug development standards and easing patient burden," Mr. Chatterjee says.
In addition to providing the technology and guidance needed to properly build and implement a synthetic control arm, Mr. Chatterjee says Medidata also has considerable regulatory expertise and regularly helps customers develop a regulatory strategy and provides counsel throughout the process.
As far as FDA acceptance goes, Mr. Chatterjee says the agency is cautiously optimistic about external control arms in general, but that their applications tend to be disease- or indication-specific. “The FDA may be more likely to accept an external control arm in single-arm trial situations where a traditional control group is unethical, or in instances where a trial may be comparing the results of their treatment to published results of another, but the patient populations described in each literature are different," he says. “However, the FDA is still wary of trial designs in which a synthetic control arm is meant to entirely replace traditional data due to concerns that synthetic data is not a one-to-one match to traditional data, which would risk inaccurate trial results and conclusions. However, we think progress is being made in accelerating FDA approvals because the Medidata SCA has been proven to be an effective tool for various study sponsors. To further support this, Medidata collaborated with the Friends of Cancer Research on two studies to show whether synthetic control arms would produce results comparable to standard control arms, and the studies demonstrated that the outcomes would have been the same regardless of data type."
Mr. Chatterjee believes the FDA may be particularly receptive to hybrid trial models where synthetic controls supplement a standard control arm instead of replacing it outright. Last year, Medicenna got the FDA nod to include a Medidata SCA in its Phase III registrational trial in recurrent glioblastoma (rGBM) as part of a hybrid external control combining Medidata SCA patients with Standard of Care patients.
“Medicenna’s FDA acceptance has gone a long way in accelerating progress of this rGBM study," he says. “Our work with Medicenna leveraged a hybrid control group, and FDA approval of these control group methods should hopefully encourage trial planners to adopt these powerful tools in the long run and open new possibilities in control group recruitment."
Thanks to leveraging this type of external control, Medicenna and Medidata have been able to drastically reduce the number of patients needed to enroll in the study to achieve the primary endpoint, meaning that more patients will receive the experimental treatment. “There are no established therapies to prolong life for people suffering with rGBM, so the hybrid SCA provides great hope for patients with this disease," Mr. Chatterjee says.
“Over and above regulatory purposes, synthetic or external controls provide context to reimbursement and pricing decisions, and we are seeing many payers/health technology assessments (HTAs) engaged in such discussions," Dr. Madison says.
Matching Up the Data
While external control arms provide many considerable advantages, the model does present some challenges.
“In terms of data reliability, a synthetic control group may not effectively replace a traditional control group because the baseline characteristics of the patient populations between the synthetic control and target trial don’t align or the results from a synthetic control group may not replicate those of a traditional one," Mr. Chatterjee says. “To circumnavigate these issues, trial planners must 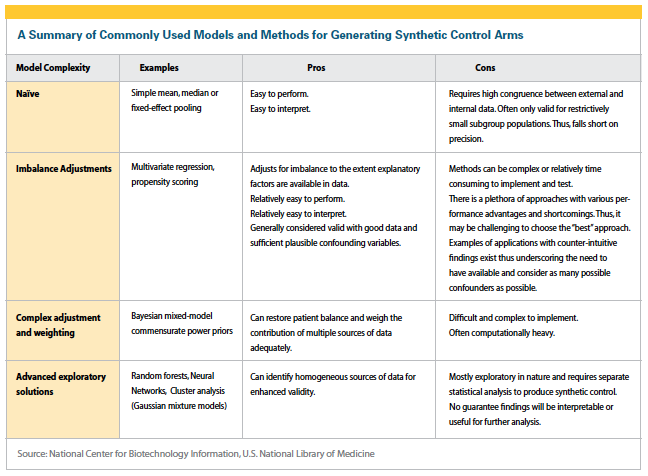 ensure they are leveraging large enough pools of high-quality, curated patient-level data that they feel confident will be on par with real-time control data. Tools like Medidata’s SCA offering will be essential in achieving this feat."
ensure they are leveraging large enough pools of high-quality, curated patient-level data that they feel confident will be on par with real-time control data. Tools like Medidata’s SCA offering will be essential in achieving this feat."
Dr. Madison says when examining the drawbacks of synthetic control arms, it’s important to consider the source they’re derived from as well as how the results will be used.
“An external control arm can range from a model to a study being derived from de-identified RWD that is quite independent of the trial arm — for example, claims or EMR data from regions with limited overlap with the trial patients — to controls derived from the same investigator sites who enrolled the trial patients and are carefully matched to each trial patient," she says. “The former may be more suitable for purposes of market access, whereas the latter can address a primary concern of ECAs — that is, the comparability of the populations in the ECA and the treatment arm."
What could make external control models more effective, but possibly less efficient, would be to have them be concurrent external controls rather than historical external controls.
“Concurrent controls are less biased, as the comparison is based on current standard of care and doesn’t need to adjust for temporal trends," Dr. Madison says. “But they can be less efficient, as the comparator arm has to accumulate data in parallel with the trial."
Despite these challenges, our experts believe use of synthetic control arms will continue to increase. Dr. Madison says Certara is already seeing an increase in requests from regulators such as the European Medicines Agency (EMA) to provide external controls for non-interventional post-authorization safety study (PASS) studies.
“As the number of complicated, targeted therapies such as cell and gene therapies grow, the use of external controls to support initial regulatory filings will likely increase, as it will become increasingly infeasible to conduct studies with internal comparators in a timely or ethical manner," she says. “I also see the use of ECAs becoming more frequent to support payer arguments and HTA decision-making."
Mr. Chatterjee envisions more trial planners adopting synthetic control arm solutions over the next 12 months, in particular gradually working in synthetic data through hybrid models.
“If the last 18 months of the pandemic have proven anything to our industry, it’s that decentralized trial designs and synthetic control arms are necessary innovations, and thanks to their proven success, are not going away anytime soon," he says. “We have seen how novel methods and capabilities have drastically accelerated bringing new treatments to market and have expanded the population of patients willing and able to participate in clinical trials. I am confident we will be seeing more use of these powerful tools in more trials going forward."(PV)